
INTRODUCTION
Asymmetric reduction of prochiral ketones to give enantiopure alcohols is an essential transformation with diverse applications in pharmaceutical as well as chemical industry. Although an extensive study has already been done in this area of asymmetric conversion, there remains always a need to explore more ecofriendly, stable, cheaper and easily available catalyst systems.
Undoubtedly, biocatalysis, metal catalysis and organocatalysis are regarded as three major pillars of modern asymmetric chemistry. Organocatalysis fascinates the synthetic organic chemist due to its easy availability, less toxicity and cost effectiveness.
The oxazaborolidine catalyst systems prepared from chiral amino alcohols and borane source have been extensively studied for the asymmetric reduction1-3. The parent catalyst H-CBZ (1) in situ synthesized from α,α-diphenylpyrrolidine methanol and BH3-THF has been found effective to furnish the asymmetric transformation, however due to its relatively less stability to air and moisture its corresponding B-methyl derivative (2) is favored.
Several other amino alcohols derived from natural and unnatural sources were then synthesized and applied as catalysts for various asymmetric transformations4. Chirally pure cis-1-amino-2-indanols have also been reported for asymmetric transformation.5,6
Didier and coworkers reported (1S, 2R)-(-)-cis-1-amino-2-indanol (3) as a chiral catalyst for asymmetric reduction of acetophenone using stoichiometric amounts of 1,3,2-oxazaborolidine B-H compounds. However the authors concluded that no system was found to be efficient with catalytic amounts of ligand (studied)6.
In order to achieve high enantioselectivity the essential requirement for chiral catalyst is, it must possess substituent at one face of transition state which can block attack from that side. Considering the structural advantages of indanyl moiety during the chiral transition state Gao Yun and coworkers studied and reported cis -1-amino-2-indanol for asymmetric reduction of aromatic prochiral ketones using BH3-THF System.7
The use of borane reagents has limitations due to difficulties in their handling, transportation and storage during commercial applications despite of their commercial availability. Periasamy and coworkers reported tetraalkylammonium borohydride, methyl iodide along with (S)-α,α-diphenylpyrrolidine methanol as an efficient catalyst system for asymmetric reduction of prochiral ketones by in situ formation of oxazaborolidine catalyst at 250C in THF to their corresponding alcohols with up to 99 % ee8. In the past, Periasamy and coworkers also reported that BH3-THF can be prepared in situ using sodium borohydride and iodine in THF. However the combination of α,α-diphenylpyrrolidine methanol , sodium borohydride and iodine gave poor results in asymmetric reduction of acetophenone. The primary reason for this fact was thought to be sparing solubility of sodium borohydride in THF9. Accordingly we have undertaken a study to employ (1S, 2R)-(-)-cis-1-amino-2-indanol as a chiral catalyst along with various borohydride reagents like sodium borohydride (4) tetramethylammonium borohydride (5) tetraethylammonium borohydride (6) and tetrabutylammonium borohydride (7).
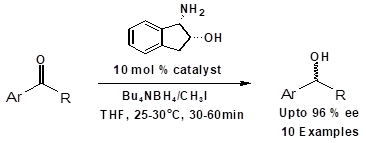
Here in we report, in situ synthesis of chiral oxazaborolidine organocatalyst by using (1S, 2R)- (-)-cis-1-amino-2-indanol, tetrabutyl-ammonium borohydride and methyl iodide to reduce prochiral ketones to the corresponding alcohols with enantiomeric excess up to 96 %.
MATERIALS AND METHOD
General Information
All reactions were carried out under inert atmosphere. Thin layer chromatography (TLC) was performed on precoated aluminium TLC plates, with detection by ninhydrin stain. Products were purified by column chromatography (Ethyl acetate and Hexane solvent system).The products were identified by spectral data (1H NMR, 13C NMR, IR and physical constants) and compared with literature values.
Enantiomeric excess was determined by HPLC using chiral column, Chiralcel-OD-H. Absolute configuration was assigned by comparison of sign of the specific rotation with that of a literature value.
The physical constants were determined using digital melting point apparatus and were observed to be uncorrected. Spectral analysis was performed by 1H-NMR spectra recorded in DMSO-d6 and CDCl3 on a Bruker Avance III, 400 MHz and Bruker Avance 300 MHz. Chemical shifts were reported as d (ppm) scale using TMS as internal standard with multiplicities and number of protons. Infrared spectra were recorded on a Perkin Elmer IR Spectrometer with nmax value reported in cm-1.
All the products obtained and discussed in this work have been reported and characterized by suitable technique such as 1H NMR, 13C NMR, IR and were compared with previously reported data.
General experimental procedure for asymmetric reduction of ketones using (1S, 2R)-(-)-cis-1-amino-2-indanol, tetrabutyl-ammonium borohydride and methyl iodide
Tetrabutylammonium borohydride (7) (5 mmol) and (1S, 2R)-(-)-cis-1-amino-2-indanol (10 mol %) in THF (5 times) were taken in a three neck RB flask. The contents were stirred at 25 -300C for about 10 min under nitrogen atmosphere. Methyl iodide (5 mmol) was added using a syringe and the reaction mixture was stirred for about 30 min. Acetophenone derivative (8a-8k) (5 mmol) in THF (5 times) was added drop wisely for about 30 min under nitrogen atmosphere. The reaction mixture was stirred till reaction completion. The mixture was carefully quenched with HCl to get pH 5.0-6.0, the organic layer was extracted with Dichoromethane (10 times). The combined organic extract was washed with brine (3 Times), dried over anhydrous Na2SO4, and the solvent was evaporated to give residue. The residue was purified on a silica gel column using hexane/ethyl acetate as eluent to furnish desired chirally pure alcohol (9a-9k).
RESULTS AND DISCUSSION
Initially various borohydride reagents like sodium borohydride (4) tetramethylammonium borohydride (5) tetraethylammonium borohydride (6) and tetrabutylammonium borohydride (7) were examined (Figure 2) by employing these reagents for the asymmetric reduction of acetophenone using (1S, 2R)-(-)-cis-1-amino-2-indanol 3 as a chiral catalyst as per the reported process, Scheme 1.



Scheme 1: Reduction of acetophenone using in situ generated oxazaborolidine catalyst
It was observed that acetophenone when subjected for asymmetric reduction by using (1S, 2R)- (-)-cis-1-amino-2-indanol (3) as a chiral catalyst with sodium borohydride and methyl iodide gave very poor enantioselectivity. Comparatively better enantioselectivity (67-73%) was obtained with tetramethylammonium borohydride (5) tetraethylammonium borohydride (6) however longer reaction time required. Employment of tetrabutylammonium borohydride (7) and methyl iodide with chiral organocatalyst (3) resulted in even better enantioselectivity (91%) with 89 % yield. The improved enantioselectivity for reagent (7) attributed to better solubility of tetrabutyl ammonium borohydride (7) in THF.
It was also observed that replacing the reaction solvent from THF to DCM or employing iodine instead of methyl iodide lowers the enantioselectivity. Initially we carried out reactions with 5 mol % of catalyst (3). When catalyst loading was increased to 10 mol %, enantiomeric excess increased upto 93 %. However further increase in catalyst loading (20 mol %) could not enhance enantioselectivity. The enantioselective reduction of acetophenone under different conditions is given in Table 1.
Table 1: Enantioselective reduction of acetophenone under different conditionsa
| S. No. | Catalayst
Loading (mol %) |
Reagents | Solvent | Yield
(%)b |
Enantiomeric Excessc | Absolute Configurationd |
| 1 | 5 | 4 / CH3I | THF | 85 | 63 | (R) |
| 2 | 5 | 5 / CH3I | THF | 81 | 67 | (R) |
| 3 | 5 | 6 / CH3I | THF | 85 | 73 | (R) |
| 4 | 5 | 7 / CH3I | THF | 89 | 91 | (R) |
| 5 | 5 | 7 / Iodine | THF | 87 | 85 | (R) |
| 6 | 5 | 7 / CH3I | DCM | 86 | 84 | (R) |
| 7 | 10 | 7 / CH3I | THF | 92 | 93 | (R) |
| 8 | 20 | 7 / CH3I | THF | 89 | 93 | (R) |
- All reactions were carried out using 5 mmol of borohydride reagent, 5 mmol of methyl iodide, 5 mmol of acetophenone, 5 – 20 mol % of (1S, 2R)-(-)-cis-1-amino-2-indanol in 5 times of solvent.
- The yields are of isolated product after purification by column chromatography. The products were identified by spectral data (1H NMR, 13C NMR, IR and physical constants) and compared with literature values.
- Enantiomeric excess was determined by HPLC using chiral column, Chiralcel-OD-H
- Absolute configuration was assigned by comparison of sign of the specific rotation with that of a literature value.
The substrate scope of this protocol is studied by varying substituent on aromatic ring of acetophenone, Table 2. Better enantioselectivity was obtained with acetophenone derivatives bearing electron withdrawing groups compared to electron donating substituents.
Table 2: Substrate scope for asymmetric reduction of ketonesa



- All reactions were carried out using 5 mmol of tetrabutylammonium borohydride, 5 mmol of methyl iodide, 5 mmol of acetophenone and 10 mol % of (1S,2R)-(-)-cis-1-amino-2-indanol in 5 times THF.
- The yields are of isolated product after purification by column chromatography. The products were identified by spectral data (1H NMR, 13C NMR, IR, and physical constants) and compared with literature values.
- Enantiomeric excess was determined by HPLC using chiral column, Chiralcel-OD-H
- Absolute configuration was assigned by comparison of sign of the specific rotation with that of a literature value.
a. All reactions were carried out using 5 mmol of tetrabutylammonium borohydride, 5 mmol of methyl iodide, 5 mmol of acetophenone and 10 mol % of (1S,2R)-(-)-cis-1-amino-2-indanol in 5 times THF. b. The yields are of isolated product after purification by column chromatography. The products were identified by spectral data (1H NMR, 13C NMR, IR, and physical constants) and compared with literature values. c. Enantiomeric excess was determined by HPLC using chiral column, Chiralcel-OD-H d. Absolute configuration was assigned by comparison of sign of the specific rotation with that of a literature value. |
CONCLUSION
In conclusion, we have explored in situ synthesis of chiral oxazaborolidine catalyst by using (1S, 2R)-(-)-cis-1-amino-2-indanol, tetrabutylammonium borohydride and methyl iodide. Various borane reduction systems have been studied and reported in this work. The chiral organocatalyst (1S,2R)-(-)-cis-1-amino-2-indanol along with tetrabutylammonium bromide, methyl iodide was employed for the reduction of a number of ortho, meta and para substituted acetophenones. It was inferred that acetophenone derivatives bearing electron withdrawing groups depicted better enantioselectivity compared to electron donating substituents.
ACKNOWLEDGEMENT
We are thankful to the analytical department of Sun Pharmaceutical Industries Limited, Gurgaon, India for their characterization
analysis support.
REFERENCES
- (a) Singh, V. K. (1992). Practical and useful methods for the enantioselective reduction of unsymmetrical ketones. Synthesis, 1992(7), 605-617. https://doi.org/10.1055/s-1992-26174
(b) Corey, E. J., & Helal, C. J. (1998). Reduction of carbonyl compounds with chiral oxazaborolidine catalysts: a new paradigm for enantioselective catalysis and a powerful new synthetic method. Angewandte Chemie International Edition, 37(15), 1986-2012. https://doi.org/10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z
(c) Mathre, D. J., Thompson, A. S., Douglas, A. W., Hoogsteen, K., Carroll, J. D., Corley, E. G., & Grabowski, E. J. (1993). A practical process for the preparation of tetrahydro-1-methyl-3, 3-diphenyl-1H, 3H-pyrrolo [1, 2-c][1, 3, 2] oxazaborole-borane. A highly enantioselective stoichiometric and catalytic reducing agent. The Journal of Organic Chemistry, 58(10), 2880-2888. https://doi.org/10.1021/jo00062a037
- (a) Corey, E. J., Bakshi, R. K., & Shibata, S. (1987). Highly enantioselective borane reduction of ketones catalyzed by chiral oxazaborolidines. Mechanism and synthetic implications. Journal of the American Chemical Society, 109(18), 5551-5553. https://doi.org/10.1021/ja00252a056
(b) Corey, E. J., Bakshi, R. K., Shibata, S., Chen, C. P., & Singh, V. K. (1987). A stable and easily prepared catalyst for the enantioselective reduction of ketones. Applications to multistep syntheses. Journal of the American Chemical Society, 109(25), 7925-7926. https://doi.org/10.1021/ja00259a075
- (a) Itsuno, S., Ito, K., Hirao, A., & Nakahama, S. (1983). Asymmetric reduction of aromatic ketones with the reagent prepared from (S)-(–)-2-amino-3-methyl-1, 1-diphenylbutan-1-ol and borane. Journal of the Chemical Society, Chemical Communications, (8), 469-470. https://doi.org/10.1039/C39830000469
(b) Corey, E. J. (1990). New enantioselective routes to biologically interesting compounds. Pure and applied chemistry, 62(7), 1209-1216. https://doi.org/10.1351/pac199062071209
- Reddy, U. V. S., Chennapuram, M., Seki, C., Kwon, E., Okuyama, Y., & Nakano, H. (2016). Catalytic Efficiency of Primary β‐Amino Alcohols and Their Derivatives in Organocatalysis. European Journal of Organic Chemistry, 2016(24), 4124-4143. https://doi.org/10.1002/ejoc.201600164
- Thompson, W. J., Fitzgerald, P. M.,
Holloway, M. K., Emini, E. A., Darke, P. L., McKeever, B. M., … & Zugay, J. A. (1992). Synthesis and antiviral activity of a series of HIV-1 protease inhibitors with functionality tethered to the P1 or P1’phenyl design. Journal of medicinal chemistry, 35(10), 1685-1701. https://doi.org/10.1021/jm00088a003, PMid:1588551
- Didier, E., Loubinoux, B., Tombo, G. H. R., & Rihs, G. (1991). Chemo-enzymatic synthesis of 1, 2-and 1, 3-amino-alcohols and their use in the enantioselective reduction of acetophenone and anti-acetophenone oxime methyl ether with borane. Tetrahedron, 47(27), 4941-4958. https://doi.org/10.1016/S0040-4020(01)80959-5
- Hong, Y., Gao, Y., Nie, X., & Zepp, C. M. (1994). cis-1-amino-2-indanol in asymmetric synthesis. Part I. A practical catalyst system for the enantioselective borane reduction of aromatic ketones. Tetrahedron letters, 35(36), 6631-6634. https://doi.org/10.1016/S0040-4039(00)73453-8
- Anwar, S., & Periasamy, M. (2006). A convenient method for the preparation of oxazaborolidine catalyst in situ using (S)-α, α-diphenylpyrrolidinemethanol, tetrabutylammonium borohydride, and methyl iodide for the asymmetric reduction of prochiral ketones. Tetrahedron: Asymmetry, 17(23), 3244-3247. https://doi.org/10.1016/j.tetasy.2006.11.032
- Periasamy, M., Kanth, J. B., & Prasad, A. B. (1994). Convenient procedures for the asymmetric reductions utilizing α, α-diphenyl-pyrrolidinemethanol and borane complexes generated using the I2/NaBH4 system. Tetrahedron, 50(21), 6411-6416. https://doi.org/10.1016/S0040-4020(01)80657-8
1 thought on “Application of In Situ Generated Chiral Oxazaborolidine Catalyst for the Enantioselective Reduction of Prochiral Ketones”